Abstract
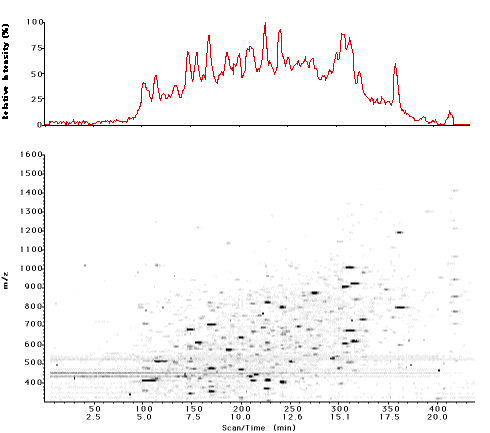
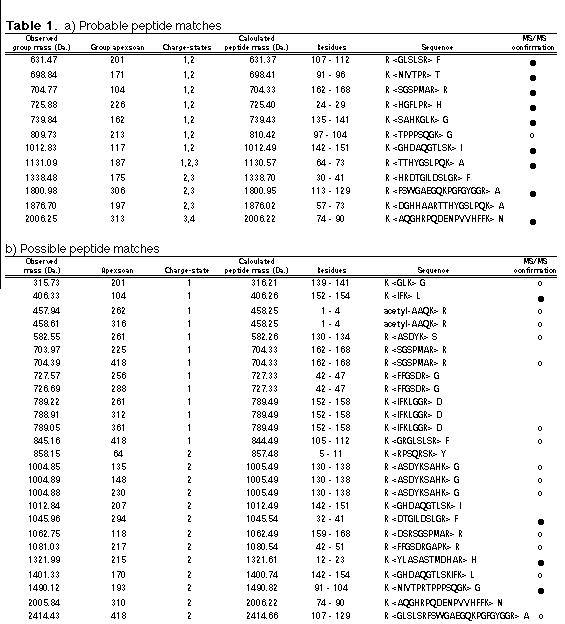
A program, Sherpa, has been developed which provides a battery of searches to allow an investigator to interpret more efficiently LC/MS and MS/MS data from protein digests by facilitating first a distinction between signal and noise, and then comparisons of observed data with peptides predicted from proteins of know sequence. However, as virtually all proteins are post-translationally modified, more is needed than just a simple matching of the data to the protein sequence. Sherpa contains specific search functions for phosphopeptides and glycopeptides. Both searches utilize ion groups and chromatographic tolerances to improve their accuracy. A built-in rating system for the results of searches draws attention to the most likely interpretations for further evaluation without excluding more remote possibilities from consideration. The flexible, nonlinear structure of Sherpa and its simple user interface allows hypotheses to be quickly tested under different settings and tolerances. As a result, the amount of manual interpretation needed is greatly reduced and focused.
 Poster Page 2
Poster Page 3
Univ. of Washington
Poster Page 2
Poster Page 3
Univ. of Washington
 Last Updated April 12, 1996
Last Updated April 12, 1996